Next: 29.6.2 Extended domains
Up: 29.6 Options for selection
Previous: 29.6 Options for selection
Standard domains are always determined first. They are used to define strong, close, weak,
and distant pairs. More accurate results can be obtained with extended domains, as described
in section 29.6.2.
- THRBP=value
- Threshold for selecting the atoms contributing to orbital domains
using the method of Boughton and Pulay (BP). As many atoms as needed to fulfill the BP criterion
are included in a domain. The order in which atoms are considered depends on the parameter MAXBP,
see below. The default is THRBP=0.98. THRBP=1.0 includes all atoms into each orbital domain,
i.e., leads to full domains. If no pairs are neglected, this should yield the canonical MP2 energy.
The criterion is somewhat basis dependent. See
section 29.9.4 for recommended values of this threshold.
- CHGMIN=value
- determines the minimum allowed Mulliken (or Löwdin) charge
for an atom (except H) in a domain, i.e., atoms with a smaller (absolute) charge are
not included, even if the THRBP criterion is not fulfilled (default 0.01).
- CHGMINH=value
- as CHGMIN, but used for H-atoms (default 0.03).
- CHGMAX=value
- If the atomic charge is larger than this value, the atom is
included, independent of any ranking.
- MAXBP=maxbp
- If maxbp=1, the atoms are ranked according to their
contribution to the Boughton-Pulay overlap. If maxbp=0 (default), the atoms are
ranked according to atomic charges.
In both cases atoms with charges greater than CHGMAX are always included, and atoms
with the same charges are added as groups.
- MULLIKEN=option
- Determines the method to determine atomic charges. MULLIKEN=0 (default):
squares of diagonal elements of
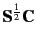 are used (Löwdin charges); MULLIKEN=1: Mulliken gross
charges are used. The first choice is less basis set dependent and more reliable with diffuse basis sets.
are used (Löwdin charges); MULLIKEN=1: Mulliken gross
charges are used. The first choice is less basis set dependent and more reliable with diffuse basis sets.
- MERGEDOM=number
- If number is greater than zero, all orbital domains containing
number or more atoms in common are merged (number=1 is treated as number=2, default 0).
This is particularly useful for geometry optimizations of conjugated or aromatic systems like, e.g., benzene.
In the latter case, MERGEDOM=1 causes the generation of full
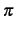 -domains, i.e., the
domains for all three -orbitals comprise all carbon basis functions. Note that
the merged domains are generated after the above print of orbital domains, and
information about merged domains is printed separately.
See section 29.9.7 for further discussion of geometry optimizations.
-domains, i.e., the
domains for all three -orbitals comprise all carbon basis functions. Note that
the merged domains are generated after the above print of orbital domains, and
information about merged domains is printed separately.
See section 29.9.7 for further discussion of geometry optimizations.
There are some other options which should normally not be modified:
- DELBAS=ibaso
- This parameter determines the method for eliminating redundant functions of
pair domains.
ibaso=0: The space of normalized eigenvectors of
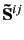 , which correspond to small
eigenvalues, is eliminated (default). Any other value is not recommended and not further documented.
, which correspond to small
eigenvalues, is eliminated (default). Any other value is not recommended and not further documented.
- DELCOR=nshell
- Activates elimination of basis functions corresponding to core orbitals.
If nshell=1, only
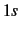 -functions are eliminated from projected space.
If nshell=2 (default) functions on first-row atoms, and
,
-functions are eliminated from projected space.
If nshell=2 (default) functions on first-row atoms, and
, 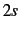 , and
, and 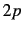 -functions are eliminated on second-row atoms. Nothing is eliminated on H or He
atoms. If effective core potentials are used, nothing is deleted at the corresponding atom. Also,
functions are only deleted if the norm of the projected function is below THRCOR (default 0.1)
-functions are eliminated on second-row atoms. Nothing is eliminated on H or He
atoms. If effective core potentials are used, nothing is deleted at the corresponding atom. Also,
functions are only deleted if the norm of the projected function is below THRCOR (default 0.1)
Next: 29.6.2 Extended domains
Up: 29.6 Options for selection
Previous: 29.6 Options for selection
molpro@molpro.net
Sep 24, 2008