Next: 39.2.1 Choice of coordinates
Up: 39 ENERGY GRADIENTS
Previous: 39.1.9 Example
39.2 Numerical gradients
It is possible to compute gradients by finite differences using
FORCE,NUMERICAL,options
Numerical gradients are computed automatically if no analytical gradients are available
for the last energy calculation. By default, no further input are needed, and the
gradient will be computed for the last energy calculation. The following options
can be given on the FORCE command or on subsequent directives (see subsequent sections):
- STARTCMD=command
- The input between command and the
current FORCE command defines the energy calculation for which the gradient is computed. This input section
is executed for each displacement.
- PROC=procname
- specifies a procedure to be executed for each displacement. This must
define a complete energy calculation and must not contain gradient or Hessian calculations.
- VARIABLE=varname
- Compute the gradient of the value of variable varname. This implies numerical gradients.
The variable must be set in the corresponding energy calculation.
- COORD=ZMAT|CART|3N
- coordinates with respect to which the gradient is evaluated. See section 39.2.1 for more information.
- DISPLACE=ZMAT|SYM|UNIQUE|CART
-
Displacement coordinates to be used for numerical gradient. The default
is ZMAT if the geometry is given as a zmatrix which depends on variables, and SYM (symmetrical displacement coordinates)
otherwise. See section 39.2.1 for more information.
- SYMMETRY=AUTO|NOSYM
- Symmetry to be used in wavefunction calculations of numerical gradients.
This option is only relevant if DISPLACE=UNIQUE|CART.
If AUTO is given, the maximum possible symmetry is used for each displacement. This implies that the energy is independent
of the symmetry used. Note that this often not the case in MRCI or CASPT2 calculations. The option can also not
be used in local correlation calculations.
- AUTO
- (logical). Same as SYMMETRY=AUTO
- ZMAT
- (logical). Same as COORD=ZMAT
- OPT3N
- (logical). Same as COORD=3N
- RSTEP=rstep
- Step length for distances in numerical gradient calculations (in bohr). The default is 0.01.
- DSTEP=dstep
- Step length for symmetrical displacements (in bohr). The default is 0.01.
- ASTEP=astep
- Step length for angles in numerical gradient calculations (in degree). The default is 1.
- CENTRAL
- (logical). Use 2-point central formula; needs
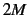 energy calculations for
energy calculations for 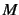 degrees of freedom.
degrees of freedom.
- FORWARD
- (logical). Use forward gradients (needs only
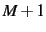 energy calculations, but less accurate)
energy calculations, but less accurate)
- FOURPOINT
- (logical). Use 4-point formula for accurate numerical gradient; needs
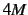 energy calculations.
energy calculations.
- NUMERICAL
- (logical). Force the use of numerical gradients, even if gradients are available.
- VARSAV
- (logical). Save gradient in variables GRADX, GRADY, GRADZ.
Example
hf
ccsd(t)
forces,numerical
The program will then automatically repeat HF and CCSD(T) at as many
geometries as needed for evaluating the gradient.
This is equivalent to
hf
ccsd(t)
forces,numerical,startcmd=hf
or, using a procedure
forces,numerical,proc=runccsdt
...
runccsdt={
hf
ccsd(t)}
Subsections
Next: 39.2.1 Choice of coordinates
Up: 39 ENERGY GRADIENTS
Previous: 39.1.9 Example
molpro@molpro.net
Sep 24, 2008