Next: 40.2.8 Hessian elements (HESSELEM)
Up: 40.2 Directives for OPTG
Previous: 40.2.6 Hessian approximations (HESSIAN)
40.2.7 Numerical Hessian (NUMHESS)
NUMHESS,options
or
NUMHESS,hstep,options
If this directive is present a numerical Hessian is computed using
finite differences. If analytical gradients are available,
one can use forward gradient differences (needs one gradient
calculation for each coordinate) or central differences
(more accurate, needs two gradient calculations for each coordinate).
For transition state optimizations it is usually sufficient
to use forward differences. If analytical gradients are not
available for the optimized method, the energy is differentiated twice.
In this case only central differences are possible.
The following options can be given:
- HSTEP=hstep
- hstep=-1: Don't calculate numerical hessian (default for minimization);
hstep=0 Calculate numerical hessian only once at the start of the optimization (default for transition state searches).
hstep=n Calculate numerical hessian after each n optimization steps.
This is useful for difficult transition state optimizations (e.g. if the eigenvalue structure of the hessian changes during the optimization).
- FORWARD
- Use forward differences (default).
- CENTRAL
- Use the more accurate central differences.
- RSTEP=rstep
- Step length for distances (in bohr). The default is 0.01.
- ASTEP=astep
- Step length for angles (in degree). The default is 0.5 or 1 for angles below and above 90 degree, respectively.
- DSTEP=dstep
- Step length for symmetrical displacements (in bohr). The default is 0.01.
- VARIABLE=varname
- Use given variable for numerical calculation of the Hessian. Note that
this disables the use of gradients, and Hessian evaluation can be very expensive.
- PROCEDURE=procname
- Procedure to be used for computing Hessian.
This procedure must be
define a complete energy calculation (orbital optimization and correlation treatment). A different method
can be used than for the optimized energy. For instance, an MP2 hessian can be used for CCSD(T) optimizations, or
a CASPT2 hessian for MRCI optimizations. By default, the same procedure is used for the hessian as for the optimized energy.
- DISPLACE=type
- type can be one of the following:
2cm
- SYMM
- Use symmetric displacement coordinates (default). This is the
only recommended option.
- CART
- Use
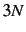 cartesian displacements (not recommended). This requires many more energy
calculations than necessary and does not preserve the molecular symmetry.
cartesian displacements (not recommended). This requires many more energy
calculations than necessary and does not preserve the molecular symmetry.
- UNIQUE
- Use symmetry-unique cartesian displacements (not recommended)
Note that the displacement type for gradient and hessian must be the same.
- CALC=icalc
- icalc=0: Recalculate the complete Hessian matrix numerically after each hstep optimization steps (default).
icalc=1: Recalculate selected Hessian matrix elements if the relative deviation of this element before and after
update (see UPDATE, section 40.2.9) is larger than thresh.
If thresh is not specified, a default value of 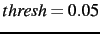 (i.e. a maximum deviation of
(i.e. a maximum deviation of 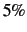 ) is used.
) is used.
icalc=2: Recalculate complete Hessian matrix if the RMS deviation of the Hessian matrix before and after update is larger than  .
If thresh is not specified a default value of
.
If thresh is not specified a default value of
- THRESH=thresh
- Threshold for partial or dynamical update of hessian, see above
Next: 40.2.8 Hessian elements (HESSELEM)
Up: 40.2 Directives for OPTG
Previous: 40.2.6 Hessian approximations (HESSIAN)
molpro@molpro.net
Sep 24, 2008