Next: 40.2.11 Transition state (saddle
Up: 40.2 Directives for OPTG
Previous: 40.2.9 Hessian update (UPDATE)
NUMERICAL,options,active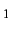 =step, active
=step, active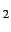 =step ...;
=step ...;
With this directive the gradients are computed by finite differences.
step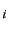 is the increment for the active geometry parameter
is the increment for the active geometry parameter 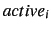 .
For active parameters which are not specified, the default values are used.
By default, the increment is 0.01 bohr for bond distances and 0.5 or 1 degree
for angles less than or greater than 90 degrees, respectively.
These defaults can be modified by specifying
RSTEP or ASTEP. DSTEP is the length of symmetrical displacements, which are used
if the optimization is performed in 3N coordinates.
.
For active parameters which are not specified, the default values are used.
By default, the increment is 0.01 bohr for bond distances and 0.5 or 1 degree
for angles less than or greater than 90 degrees, respectively.
These defaults can be modified by specifying
RSTEP or ASTEP. DSTEP is the length of symmetrical displacements, which are used
if the optimization is performed in 3N coordinates.
For each active variable, two energy calculations are
necessary in each geometry optimization step - so numerical optimizations may be
expensive! In optimizations of 3N coordinates symmetrical displacement coordinates are normally used
to minimize the number of energy calculations.
(see section 39.2.1).
For optimization of special energies see VARIABLE section 40.2.17.
The following options can be given:
- RSTEP=rstep
- Step length for distances (in bohr). The default is 0.01.
- ASTEP=astep
- Step length for angles (in degree). The default is 0.5 or 1 for angles below and above 90 degree, respectively.
- DSTEP=dstep
- Step length for symmetrical displacements (in bohr). The default is 0.01.
- CENTRAL
- Use central differences for gradient (default)
- FORWARD
- Use forward differences (not recommended for gradient).
- FOURPOINT
- Use four-point formula for very accurate numerical gradients.
- PROCEDURE=procname
- Use given procedure for numerical calculation of the gradient. This procedure must define
a complete energy calculation (orbital optimization and correlation treatment).
- VARIABLE=varname
- Use given variable for numerical calculation of the gradient.
- DISPLACE=type
- The displacement type. Note that the displacement type for gradient and hessian must be the same.
type can be one of the following:
2cm
- SYMM
- Use symmetric displacement coordinates (default). This is the
only recommended option.
- CART
- Use
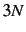 cartesian displacements (not recommended). This requires many more energy
calculations than necessary and does not preserve the molecular symmetry.
cartesian displacements (not recommended). This requires many more energy
calculations than necessary and does not preserve the molecular symmetry.
- UNIQUE
- Use symmetry-unique cartesian displacements (not recommended)
Next: 40.2.11 Transition state (saddle
Up: 40.2 Directives for OPTG
Previous: 40.2.9 Hessian update (UPDATE)
molpro@molpro.net
Sep 24, 2008