Next: 44.2 Standard Problems
Up: 44 THE VSCF PROGRAM
Previous: 44 THE VSCF PROGRAM
The following options are available:
- PMP=value
- Vibrational angular momentum terms (Coriolis coupling), i.e.
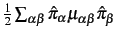 ,
and the Watson correction term are by default switched off.
PMP=1 adds the Watson correction term (see eq. 20) as a
pseudo-potential like contribution to the fine grid of the potential. PMP=2 allows for the
calculation of the integrals of the PMP operator using the approximation that the
,
and the Watson correction term are by default switched off.
PMP=1 adds the Watson correction term (see eq. 20) as a
pseudo-potential like contribution to the fine grid of the potential. PMP=2 allows for the
calculation of the integrals of the PMP operator using the approximation that the 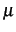 tensor
is given as the inverse of the moment of inertia tensor at equilibrium geometry.
tensor
is given as the inverse of the moment of inertia tensor at equilibrium geometry.
- COMBI=value
- By default the VSCF program calculates the fundamental modes of the molecule only.
However, choosing COMBI=1 allows
for the calculation of the first vibrational overtones and
 combination bands consisting of
two modes in the first vibrational level.
combination bands consisting of
two modes in the first vibrational level.
- SOLVER=value
- For solving the one-dimensional Schrödinger equation two different algorithms can be used.
The default, i.e. SOLVER=1, calls the discrete variable representation (DVR) as proposed
by Hamilton and Light. Alternatively, the collocation algorithm of Young and Peet can be used (SOLVER=2).
- THERMO=value
- THERMO=1 allows for the improved calculation of thermodynamical quantities
(compare the THERMO keyword in combination with a harmonic frequency calculation). However,
the approach used here is an approximation: While the harmonic approximation ist still retained in the
equation for the partition functions, the actual values of the frequencies entering
into these functions are the anharmonic values derived from the VSCF calculation.
- ROTJ=value
- Rovibrational levels can be computed in an approximative fashion only
(this does not work in combination with
the COMBI option). Once the VSCF equations have been solved, the rotational constants will be
computed from the vibrationally averaged structures for each vibrational level. This allows for a rough estimate
and very fast calculation of the rovibrational levels. ROTJ=
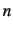 determines the value of
determines the value of 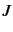 . A negative
number for results in a calculation of all rovibrational levels from
. A negative
number for results in a calculation of all rovibrational levels from 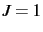 up to the specified
value.
up to the specified
value.
- DIPOLE=value
- DIPOLE=1 allows for the calculation of infrared intensities.
Calculation of infrared intensities requires the calculation of dipole surfaces within the SURF
program. By default intensities will not be computed.
- NDIM=value
- The expansion of the potential in the VSCF calculation can differ from the
expansion in the SURF calculation. However, only values less or equal to the one used
in the surface calculation can be used.
- INFO=value
- INFO=1 provides a list of the values of all relevant program parameters (options).
Next: 44.2 Standard Problems
Up: 44 THE VSCF PROGRAM
Previous: 44 THE VSCF PROGRAM
molpro@molpro.net
Sep 24, 2008