Next: 45.2 Recommendations
Up: 45 THE VCI PROGRAM
Previous: 45 THE VCI PROGRAM
The following options are available:
- VERSION=value
- The default (i.e. VERSION=1) is a configuration selective VCI implementation, which is the fastest
VCI code available in MOLPRO. The configuration selection is based on the vibrational ground state,
while the final VCI matrix is state-specific. This approximation reduces the computational effort within
the screening.
VERSION=2 is a state-specific configuration selective VCI implementation, which usually is more
reliable - but also more expensive - than VERSION=1. However, the configuration selection still
introduces some inaccuracies which may
not be desirable for very accurate calculations. A small correction to the state-specific configuration
selection VCI may be
invoked by choosing VERSION=3. This may increase the CPU-time by a factor of two.
All configuration selective VCI programs have been parallelized in a fashion, that
the individual columns of the VCI matrices are send to different processors. A conventional
VCI calculation can be performed by choosing VERSION=4. This version yields the most accurate results.
However, in this case the VCI matrix can be
huge and usually these calculations are limited either by the CPU demands for diagonalizing the VCI matrix
or by the corresponding memory requests.
- CITYPE=value
- CTYPE defines the maximum number of simultaneous excitations,
i.e. Singles, Doubles, Triples, ... and
thus determines the kind of calculations, i.e. VCISD, VCISDT, ... The default is CITYPE=4 (VCISDTQ),
which appears to be a fair compromise between accuracy and computational speed.
The maximum excitation level is currently limited to CITYPE=6.
- LEVEX=value
- LEVEX determines the level of excitation within one mode, i.e.
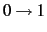 ,
,
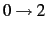 ,
,
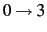 , ... The default is LEVEX=4, which was found to be sufficient for many applications.
, ... The default is LEVEX=4, which was found to be sufficient for many applications.
- CIMAX=value
- CIMAX is the maximum excitation level corresponding to CITYPE and LEVEX. In principle,
a triple configuration
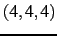 would contribute to the VCI space. However, CIMAX=7 restricts this
to
would contribute to the VCI space. However, CIMAX=7 restricts this
to 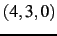 ,
, 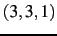 ,
, 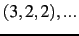 . The default is CIMAX=8, which needs to be extended for certain applications.
. The default is CIMAX=8, which needs to be extended for certain applications.
- CITHR=value
- CITHR controls the threshold within the configuration selection scheme
(see VERSION=1-3). The default is 0.99995.
- BSIZE=value
- The size of the screening blocks within the configuration selection algorithm
is measured in terms of the number of single and double excitations, i.e. BSIZE=1.0. The default is
BSIZE=1.5 (for details see the reference given above).
- PMP=value
- Vibrational angular momentum terms (Coriolis coupling), i.e.
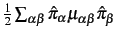 ,
and the Watson correction term are by default switched off.
PMP=1 adds the Watson correction term (see eq. 20) as a
pseudo-potential like contribution to the fine grid of the potential. PMP=2 allows for the
calculation of the integrals of the PMP operator using the approximation that the
,
and the Watson correction term are by default switched off.
PMP=1 adds the Watson correction term (see eq. 20) as a
pseudo-potential like contribution to the fine grid of the potential. PMP=2 allows for the
calculation of the integrals of the PMP operator using the approximation that the 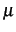 tensor
is given as the inverse of the moment of inertia tensor at equilibrium geometry. The PMP=2
option includes diagonal contributions in the VCI matrix only. This is significantly faster
than calculating the contributions for all matrix elements (which corresponds to PMP=3
and usually introduces only small deviations.
tensor
is given as the inverse of the moment of inertia tensor at equilibrium geometry. The PMP=2
option includes diagonal contributions in the VCI matrix only. This is significantly faster
than calculating the contributions for all matrix elements (which corresponds to PMP=3
and usually introduces only small deviations.
- COMBI=value
- By default the VCI program calculates the fundamental modes of the molecule only.
However, choosing COMBI=1 allows
for the calculation of the first vibrational overtones and
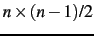 combination bands consisting of
two modes in the first vibrational level.
combination bands consisting of
two modes in the first vibrational level.
- THERMO=value
- THERMO=1 allows for the improved calculation of thermodynamical quantities
(compare the THERMO keyword in combination with a harmonic frequency calculation). However,
the approach used here is an approximation: While the harmonic approximation ist still retained in the
equation for the partition functions, the actual values of the frequencies entering
into these functions are the anharmonic values derived from the VCI calculation.
- ROTJ=value
- Rovibrational levels can be computed in an approximative fashion only
(this does not work in combination with
the COMBI option). Once the VCI wave function has been determined, the rotational constants will be
computed from the vibrationally averaged structures for each vibrational level. This allows for a rough estimate
and very fast calculation of the rovibrational levels. ROTJ=
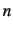 determines the value of
determines the value of 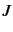 . A negative
number for results in a calculation of all rovibrational levels from
. A negative
number for results in a calculation of all rovibrational levels from 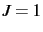 up to the specified
value.
up to the specified
value.
- DIPOLE=value
- DIPOLE=1 allows for the calculation of infrared intensities.
Calculation of infrared intensities requires the calculation of dipole surfaces within the SURF
program. By default intensities will not be computed.
- NDIM=value
- The expansion of the potential in the VCI calculation can differ from the
expansion in the SURF calculation. However, only values less or equal to the one used
in the surface calculation can be used.
- MPG=value
- Symmetry of the molecule will be recognized automatically within the
VCI calculations. MPG=1 switches symmetry off.
- INFO=value
- INFO=1 provides a list of the values of all relevant program parameters (options).
Next: 45.2 Recommendations
Up: 45 THE VCI PROGRAM
Previous: 45 THE VCI PROGRAM
molpro@molpro.net
Sep 24, 2008