Next: 2 MOLPRO on the
Up: manual
Previous: manual
MOLPRO is a complete system of ab initio programs for
molecular electronic structure calculations, designed and maintained by
H.-J. Werner and P. J. Knowles, and containing contributions from a number
of other authors. As distinct from other commonly used quantum chemistry
packages, the emphasis is on highly accurate computations,
with extensive treatment of the electron correlation problem
through the multiconfiguration-reference CI, coupled cluster and associated
methods. Using recently developed integral-direct local electron correlation
methods, which significantly reduce the increase of the computational cost with molecular size,
accurate ab initio calculations can be performed for much larger molecules than
with most other programs.
The heart of the program consists of the multiconfiguration SCF,
multireference CI, and coupled-cluster
routines, and these are accompanied by a full set of
supporting features. The package comprises
- Integral generation for generally contracted symmetry adapted gaussian
basis functions (
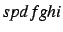 ). There are two programs with identical
functionality:
the preferred code is Seward (R. Lindh) which
is the best on most machines;
Argos (R. M. Pitzer) is available as an alternative, and in
some cases is optimum for small memory
scalar machines.
Also two different gradient integral codes, namely Cadpac (R. Amos) and
Alaska (R. Lindh) are available. Only the latter allows the use of
generally contracted symmetry adapted gaussian
basis functions.
). There are two programs with identical
functionality:
the preferred code is Seward (R. Lindh) which
is the best on most machines;
Argos (R. M. Pitzer) is available as an alternative, and in
some cases is optimum for small memory
scalar machines.
Also two different gradient integral codes, namely Cadpac (R. Amos) and
Alaska (R. Lindh) are available. Only the latter allows the use of
generally contracted symmetry adapted gaussian
basis functions.
- Effective Core Potentials (contributions from H. Stoll).
- Many one-electron properties.
- Some two-electron properties, e.g.
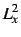 ,
, 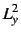 ,
, 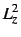 ,
, 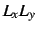 etc..
etc..
- Closed-shell and open-shell (spin restricted and unrestricted) self consistent field.
- Density-functional theory in the Kohn-Sham framework with various gradient
corrected exchange and correlation potentials.
- Multiconfiguration self consistent field. This is the quadratically
convergent MCSCF procedure described in J. Chem. Phys. 82 (1985) 5053. The
program can optimize a weighted energy average of several states, and is
capable of treating both completely general configuration expansions and
also long CASSCF expansions as described in Chem. Phys. Letters 115 (1985)
259.
- Multireference CI. As well as the usual single reference function
approaches (MP2, SDCI, CEPA), this module implements the internally
contracted multireference CI method as described in J. Chem. Phys. 89
(1988) 5803 and Chem. Phys. Lett. 145 (1988) 514. Non variational variants
(e.g. MR-ACPF), as described in Theor. Chim. Acta 78 (1990) 175, are also
available. Electronically excited states can be computed as described in
Theor. Chim. Acta, 84 95 (1992).
- Multireference second-order and third-order perturbation theory (MR-PT2, MR-PT3)
as described in Mol. Phys. 89, 645 (1996) and
J. Chem. Phys. 112, 5546 (2000).
- Møller-Plesset perturbation theory (MPPT),
Coupled-Cluster (CCSD), Quadratic configuration interaction
(QCISD), and Brueckner Coupled-Cluster (BCCD) for closed shell
systems, as described in
Chem. Phys. Lett. 190 (1992) 1.
Perturbative corrections for triple excitations can also be calculated
(Chem. Phys. Letters 227 (1994) 321).
- Open-shell coupled cluster theories as described in
J. Chem. Phys. 99 (1993) 5219,
Chem. Phys. Letters 227 (1994) 321.
- Full Configuration Interaction. This is the determinant based benchmarking
program described in Comp. Phys. Commun. 54 (1989) 75.
- Analytical energy gradients for SCF, DFT, state-averaged MCSCF/CASSCF,
MP2 and QCISD methods.
- Analytical non-adiabatic coupling matrix elements for MCSCF.
- Valence-Bond analysis of CASSCF wavefunction, and energy-optimized
valence bond wavefunctions as described in
Int. J. Quant. Chem. 65, 439 (1997).
- One-electron transition properties for MCSCF and MRCI wavefunctions.
- Spin-orbit coupling, as described in
Mol. Phys., 98, 1823 (2000).
- Some two-electron transition properties for MCSCF wavefunctions (e.g.,
etc.).
- Population analysis.
- Orbital localization.
- Distributed Multipole Analysis (A. J. Stone).
- Automatic geometry optimization as described in
J. Comp. Chem. 18, (1997), 1473.
- Automatic calculation of vibrational frequencies, intensities,
and thermodynamic properties.
- Reaction path following, as described in
Theor. Chem. Acc. 100, (1998), 21.
- Various utilities allowing other more general optimizations,
looping and branching (e.g., for automatic generation of complete
potential energy surfaces), general housekeeping operations.
- Geometry output in XYZ,
MOLDEN
and
Gaussian
formats; molecular orbital and frequency output in
MOLDEN
format.
- Integral-direct implementation of all Hartree-Fock, DFT and
pair-correlated methods (MP, CCSD, MRCI etc.), as described
in Mol. Phys., 96, (1999), 719. At present, perturbative
triple excitation methods are not implemented.
- Local second-order Møller-Plesset perturbation theory (LMP2) as
in
Chem. Phys. Lett. 290, 143 (1998),
J. Chem. Phys. 111, 5691 (1999),
and J. Chem. Phys. 113, 9443 (2000),
(and references therein).
- Analytical energy gradients for LMP2, as described in
J. Chem. Phys. 108, (1998), 5185.
- Parallel execution on distributed memory machines, as
described in J. Comp. Chem. 19, (1998), 1215.
At present, SCF, DFT, MRCI, MP2, LMP2, CCSD(T) energies and SCF, DFT
gradients are parallelized when running with conventional integral
evaluation; integral-direct SCF, DFT and LMP2 are also parallel.
The program is written mostly in standard Fortran-90. Those parts which
are machine dependent are maintained through the use of a supplied
preprocessor, which allows easy interconversion between versions for
different machines.
Each release of the program is ported and tested on
a number of
IBM RS/6000,
Hewlett-Packard,
Silicon Graphics,
Compaq,
and
Linux
systems. A fuller description of the hardware and operating systems of
these machines can be found at
http://www.tc.bham.ac.uk/molpro/machines.html.
The program additionally runs on
Cray,
Sun,
Convex, Fujitsu and NEC SX4 platforms, as well as older architectures
and/or operating systems from the primary list; however, testing is not carried out regularly
on these systems, and hand-tuning of code may be necessary on porting.
A large library of commonly used orbital
basis sets is available, which can be extended as required.
There is a comprehensive users' manual, which includes
installation instructions. The manual is available in PostScript, PDF and
also in HTML for mounting on a Worldwide Web server.
Future enhancements presently under development include
- Local coupled cluster theory (LCCSD) as described in J. Chem. Phys.
104, (1996), 6286 and J. Chem. Phys. 114, 661 (2001),
with perturbative treatment of triple excitations, as described in
Chem. Phys. Letters 318, 370 (2000) and J. Chem. Phys. 113, 9986 (2000).
- Enhancements to the efficiency of the DFT integration.
- Analytical energy gradients for CCSD, LCCSD, and CAS-PT2.
- Analytical second derivatives for SCF/MCSCF.
- Efficiency improvements for open-shell coupled cluster.
- Further parallelization.
- Open-shell MP2, LMP2 and LCCSD.
Some of these features will be included in the base version, whereas
others will be available only as optional modules. The above list is
for information only, and no representation is made that any of the
above will be available within any particular time.
Next: 2 MOLPRO on the
Up: manual
Previous: manual
P.J. Knowles and H.-J. Werner
molpro@tc.bham.ac.uk
Jan 15, 2002