Next: 23 MØLLER PLESSET PERTURBATION
Up: 22 MULTIREFERENCE RAYLEIGH SCHRÖDINGER
Previous: 22.6 Integral direct calculations
Other options can be set using the OPTION command. These options are mainly
used for testing purposes and should be used with care.
It should be noted that the only option that can be modified in the RS2C program is IFDIA:
all others only work with RS2/RS3.
OPTION,code1=value,code2=value,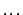
Of relevance for the CASPT2/3 program are the following options:
- IPROCS=0
- (Default). Calculation uses uncontracted singles with RS2.
- IPROCS=1
- Non-interacting singles are projected out during update. This is
an approximate procedure which should be used with care.
- IPROCS=2
- The singles are fully internally contracted in RS2. This is
achieved via a projection operator during the coefficient update and may be inefficient.
- IPROCS=3
- Only singles with one or two holes in the closed-shells are internally
contracted in RS2 using a projection operator.
- IPROCI=0
- (Default). Calculation uses uncontracted internals with RS2.
- IPROCI=1
- Internals with two holes in the inactive space are internally contracted
in RS2 using a projection operator.
- IPROCS=3,IPROCI=1
- This combination of options
reproduces with RS2 the RS2C result using projection operators. This requires lot of memory
and disk space and it is feasible only for small molecules.
- IFDIA=0
- (Default). All off-diagonal elements of the effective Fock matrix are included.
- IFDIA=1
- The internal-external block of the Fock-matrix is neglected. This eliminates the
single-pair coupling.
- IFDIA=2
- All off-diagonal elements of the Fock matrix are neglected. This corresponds
to CASPT2D of Andersson et al. Note: in this case the result is not invariant to rotations
among active orbitals!
- IHINT=0
- (Default). Only one-electron integrals are used in the zeroth-order Hamiltonian
for all interactions.
- IHINT=1
- The all-internal two-electron integrals are used in the zeroth-order Hamiltonian
for the internal-internal and single-single interactions.
- IHINT=2
- The all-internal two-electron integrals in the zeroth-order Hamiltonian
are used for the internal-internal, single-single, and pair-pair interactions. Using
IHINT=2 and IDFIA=1 corresponds to
Dyall's CAS/A method for the case that CASSCF references with no closed-shells (inactive orbitals) are used.
Note that this requires more CPU time than a standard CASPT2 calculation. Moreover,
convergence of the CAS/A method is often slow (denominator shifts specified on a SHIFT card
may be helpful in such cases). In general, we do not recommend the use of IHINT with nonzero
values.
- NOREF=1
- (Default). Interactions between reference configurations and singles
are omitted.
- NOREF=0
- Interactions between reference configurations and singles are included.
This causes a relaxation of the reference coefficients but may lead to intruder-state problems.
- IMP3=2
- After CASPT2 do variational CI using all internal configurations and the first-order
wavefunctions of all states as a basis. In this case the second-order energy will correspond to the
variational energy, and the third-order energy approximately to a Davidson-corrected energy.
This is useful in excited state calculations with near-degeneracy situations.
Next: 23 MØLLER PLESSET PERTURBATION
Up: 22 MULTIREFERENCE RAYLEIGH SCHRÖDINGER
Previous: 22.6 Integral direct calculations
P.J. Knowles and H.-J. Werner
molpro@tc.bham.ac.uk
Jan 15, 2002