Next: 34.2.4 Defining active geometry
Up: 34.2 Geometry optimization
Previous: 34.2.2 Automatic geometry optimization
METHOD,key;
key defines the optimization method.
For minimization the following options are valid for key:
- RF
- Rational Function method (default).
- AH
- Augmented Hessian method. This is similar to RF algorithm but uses a more
sophisticated step restriction algorithm.
- DIIS
- Pulay's Geometry DIIS method. As an an additional option
you may add the number of geometries to be used in GDIIS interpolation (default 5)
and the interpolation type (i.e. the subspace in which the GDIIS interpolation
is made.
METHOD,DIIS, number, type
type may be
GRAD interpolation using the gradients (default),
working good for rigid molecules,
STEP interpolation using Quasi-Newton steps which
could be advantageous in dealing with very floppy molecules,
ENER interpolation using energies, which is an intermediate between
the above two.
- QSD
- Quadratic steepest descent method of Sun and Ruedenberg.
- SRMIN
- Old version of QSD.
For transition state searches
(invoked with the ROOT option, see section 30.2.9) key can be
- RF
- Rational Function method (default).
- DIIS
- Pulay's Geometry DIIS method (see above).
- QSD
- Quadratic Steepest Descent Transition State search using
the image hessian method
(see J. Sun and K. Ruedenberg, J. Chem. Phys. 101, 2157 (1994))
The use of this option is recommended for
transition state searches - especially in complicated cases.
The optimization step is checked and the hessian is recalculated
when approaching a troublesome region of the PES.
Thus this method is somewhat safer (and often faster) in reaching convergence
than the RF or DIIS method. The hessian recalculation safeguard may be turned off
using the METHOD,QSD,NOHESS input card.
- SRTRANS
- Old version of QSD.
For reaction path following the input key is
- QSDPATH
- Quadratic Steepest Descent reaction path following.
This methods determines reaction paths (intrinsic reaction coordinates, IRCs)
by following the exact steepest descent lines of subsequent quadratic
approximations to the potential energy surface. The hessian matrix is
calculated numerically at the first optimization step and subsequently
updated by Powell or BFGS update. If a given arc length of the steepest
descent lines is exceeded, the hessian is recalculated numerically
(see OPTION section 30.2.20). For details
see J. Sun and K. Ruedenberg, J. Chem. Phys. 99, 5269 (1993)
It is also possible to recalculate the hessian after each m steps
using the NUMHES,m command (see section 30.2.18).
If the hessian matrix is recalculated in every optimization step
(NUMHES,
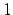 ) a algorithm different to the one
with updated hessians is used, which is very accurate. Using the
PRINT,OPT card, this algorithm prints in every optimization step
a reaction path point r which is different from the point where the
energy and the gradient is calculated but closer to the real reaction path
(for further details of the algorithm see J. Sun and K. Ruedenberg,
J. Chem. Phys. 99, 5257 (1993)).
For further input options of the QSD reaction path following see OPTION
section 30.2.20.
) a algorithm different to the one
with updated hessians is used, which is very accurate. Using the
PRINT,OPT card, this algorithm prints in every optimization step
a reaction path point r which is different from the point where the
energy and the gradient is calculated but closer to the real reaction path
(for further details of the algorithm see J. Sun and K. Ruedenberg,
J. Chem. Phys. 99, 5257 (1993)).
For further input options of the QSD reaction path following see OPTION
section 30.2.20.
- SRSTEEP
- Old Version of QSDPATH.
Next: 34.2.4 Defining active geometry
Up: 34.2 Geometry optimization
Previous: 34.2.2 Automatic geometry optimization
P.J. Knowles and H.-J. Werner
molpro@tc.bham.ac.uk
Jan 15, 2002