Next: 32.2 First example
Up: 32 SYMMETRY-ADAPTED INTERMOLECULAR PERTURBATION
Previous: 32 SYMMETRY-ADAPTED INTERMOLECULAR PERTURBATION
32.1 Introduction
The SAPT (symmetry-adapted intermolecular perturbation theory) program
calculates the total interaction energy between closed-shell molecules as a
sum of individual first and second order interaction terms, namely
electrostatic
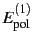 , induction
, induction
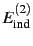 and dispersion
and dispersion
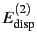 accompanied by their respective
exchange counterparts (
accompanied by their respective
exchange counterparts (
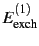 ,
,
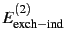 and
and
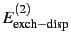 ). The latter ones arise
due to electron exchange between the monomers when the molecules are
close to each other and are sometimes denoted as Pauli repulsion.
Since all above terms are accessible through static and
(time-dependent) response density matrices of the monomers,
in principle (see section 32.4) no calculation of the dimer
wave function is required. Therefore SAPT is free from a zeroth-order basis
set superposition error which occurs in the supermolecular approach.
). The latter ones arise
due to electron exchange between the monomers when the molecules are
close to each other and are sometimes denoted as Pauli repulsion.
Since all above terms are accessible through static and
(time-dependent) response density matrices of the monomers,
in principle (see section 32.4) no calculation of the dimer
wave function is required. Therefore SAPT is free from a zeroth-order basis
set superposition error which occurs in the supermolecular approach.
References:
General Symmetry-adapted perturbation theory and many-body SAPT:
![$[1]$](img420.gif) B. Jeziorski, R. Moszynski and K. Szalewicz,
Chem. Rev. 94, 1887. (1994).
B. Jeziorski, R. Moszynski and K. Szalewicz,
Chem. Rev. 94, 1887. (1994).
DFT-SAPT:
![$[2]$](img421.gif) G. Jansen and A. Heßelmann,
J. Phys. Chem. A 105, 646 (2001).
G. Jansen and A. Heßelmann,
J. Phys. Chem. A 105, 646 (2001).
![$[3]$](img422.gif) A. Heßelmann and G. Jansen,
Chem. Phys. Lett. 357, 464 (2002).
A. Heßelmann and G. Jansen,
Chem. Phys. Lett. 357, 464 (2002).
![$[4]$](img423.gif) A. Heßelmann and G. Jansen,
Chem. Phys. Lett. 362, 319 (2002).
A. Heßelmann and G. Jansen,
Chem. Phys. Lett. 362, 319 (2002).
![$[5]$](img439.gif) A. Heßelmann and G. Jansen,
Chem. Phys. Lett. 367, 778 (2003).
A. Heßelmann and G. Jansen,
Chem. Phys. Lett. 367, 778 (2003).
![$[6]$](img440.gif) A. Heßelmann and G. Jansen,
Phys. Chem. Chem. Phys. 5, 5010 (2003).
A. Heßelmann and G. Jansen,
Phys. Chem. Chem. Phys. 5, 5010 (2003).
Density fitting DFT-SAPT (DF-DFT-SAPT):
![$[7]$](img441.gif) A. Heßelmann, G. Jansen and M. Schütz,
J. Chem. Phys. 122, 014103 (2005).
A. Heßelmann, G. Jansen and M. Schütz,
J. Chem. Phys. 122, 014103 (2005).
(See also:
K. Szalewicz, K. Patkowski and B. Jeziorski,
Struct. Bond 116, 43 (2005)
and references therein for a related approach to DFT-SAPT termed SAPT(DFT))
Next: 32.2 First example
Up: 32 SYMMETRY-ADAPTED INTERMOLECULAR PERTURBATION
Previous: 32 SYMMETRY-ADAPTED INTERMOLECULAR PERTURBATION
molpro@molpro.net
Sep 24, 2008