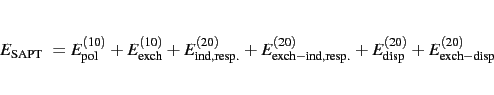![\includegraphics[scale=0.35]{he2}](img512.gif)
r=5.6
geometry={nosym; he1; he2,he1,r}
basis=avqz
!wf records
ca=2101.2
cb=2102.2
!monomer A
dummy,he2
{hf; save,$ca}
sapt;monomerA
!monomer B
dummy,he1
{hf; start,atdens; save,$cb}
sapt;monomerB
!interaction contributions
sapt;intermol,ca=$ca,cb=$cb
Here the sapt;monomerA/B store some informations about the
two monomers which are needed in the subsequent SAPT calculation
invoked by sapt;intermol. The individual interaction energy
terms are stored (in millihartree)
in distinct variables and may be collected
in arrays for producing potential energy surfaces. For example
the input
geometry={nosym; he1; he2,he1,r}
basis=avtz
!wf records
ca=2101.2
cb=2102.2
!distances
dist=[4.5,5.0,5.5,5.6,6.0,6.5,7.0]
do i=1,#dist
r=dist(i)
!monomer A
dummy,he2
{hf; save,$ca}
sapt;monomerA
!monomer B
dummy,he1
{hf; start,atdens; save,$cb}
sapt;monomerB
!interaction contributions
sapt;intermol,ca=$ca,cb=$cb
elst(i)=E1pol; exch(i)=E1ex
ind(i)=E2ind; exind(i)=E2exind
disp(i)=E2disp; exdisp(i)=E2exdisp
etot(i)=E12tot
data,truncate,$ca
enddo
{table,dist,elst,exch,ind,exind,disp,exdisp,etot
ftyp,d,d,d,d,d,d,d,d,d
plot}
yields the plot
Currently SAPT only accepts single-determinant wave functions for the monomers, i.e. from Hartree-Fock or Kohn-Sham DFT (see next section) calculations. This means that if Hartree-Fock wave functions are used for monomer, the following quantity is obtained (zero in superscript denotes that no intramonomer correlation is accounted for) [1].