Next: 32.4 High order terms
Up: 32 SYMMETRY-ADAPTED INTERMOLECULAR PERTURBATION
Previous: 32.2 First example
32.3 DFT-SAPT
It is of crucial importance to account for the intramolecular
correlation effects of the individual SAPT terms since
Hartree-Fock theory often yields poor first- and second-order
electrostatic properties. While this can be done
using many-body perturbation theory [1] (in a double perturbation theory
ansatz) a more efficient way is to use static and
time-dependent DFT theory. This variant of SAPT, termed as
DFT-SAPT [2-6], has in contrast to Hartree-Fock-SAPT the appealing
feature that the polarisation terms (
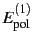 ,
,
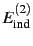 ,
,
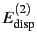 ) are
potentially exact, i.e. they come out exactly if the
exact exchange-correlation (xc) potential and the exact
(frequency-dependent) xc response kernel of the monomers
were known. On the other hand, this does not hold for the
exchange terms since Kohn-Sham theory can at best give
a good approximation to the exact density matrix of a
many-body system. It has been shown [6] that this is indeed the
the case and therefore DFT-SAPT has the potential to
produce highly accurate interaction energies comparable
to high-level supermolecular many-body perturbation or coupled cluster
theory. However, in order to achieve this accuracy, it
is of crucial importance to correct the wrong asymptotic
behaviour of the xc potential in current DFT functionals
[3-5]. This can be done by using e.g.:
) are
potentially exact, i.e. they come out exactly if the
exact exchange-correlation (xc) potential and the exact
(frequency-dependent) xc response kernel of the monomers
were known. On the other hand, this does not hold for the
exchange terms since Kohn-Sham theory can at best give
a good approximation to the exact density matrix of a
many-body system. It has been shown [6] that this is indeed the
the case and therefore DFT-SAPT has the potential to
produce highly accurate interaction energies comparable
to high-level supermolecular many-body perturbation or coupled cluster
theory. However, in order to achieve this accuracy, it
is of crucial importance to correct the wrong asymptotic
behaviour of the xc potential in current DFT functionals
[3-5]. This can be done by using e.g.:
{ks,lda; asymp,<shift>}
which activates the gradient-regulated asymptotic correction
approach of Grüning et al. (J. Chem. Phys. 114, 652 (2001))
for the respective monomer
calculation. The user has to supply a shift parameter
for the bulk potential which should approximate the
difference between the exact ionisation potential of the
monomer and the (negative) HOMO energy obtained from the
respective standard Kohn-Sham calculation. Note that this
needs to be done only once for each system.
Concerning the more technical parameters in the DFT
monomer calculations it is recommended to use
lower convergence thresholds and larger intergration grids
compared to standard Kohn-Sham calculations.
Next: 32.4 High order terms
Up: 32 SYMMETRY-ADAPTED INTERMOLECULAR PERTURBATION
Previous: 32.2 First example
molpro@molpro.net
Sep 24, 2008