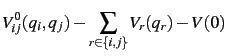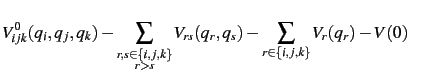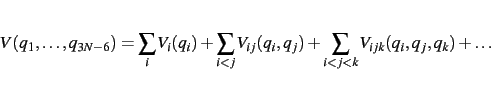 |
(15) |
The SURF program allows for the calculation of the potential energy surface
around the equilibrium structure as required for the calculation of
anharmonic frequencies (see the VSCF and VCI programs).
Currently the program is limited to the case
of one minimum. The potential is represented by energy grid points rather than
a Taylor expansion.
Within the SURF the potential energy surfaces is expanded in terms of normal coordinates.
Consequently, a harmonic frequency calculation needs to be performed first.
The potential will then be represented by a hierarchical scheme given by
label1 hf ccsd surf,start1D=label1
The SURF program is based on a iterative algorithm, i.e. grid points will be
added automatically to the grid representation of the potential until a convergence
threshold will be met. This guarantees a well-balanced description of the different
terms in the expansion of the potential and simultaneously minimizes the number of
ab initio calculations for a representation of the potential. For further details see:
G. Rauhut, Efficient Calculation of Potential Energy Surfaces for the Generation
of Vibrational Wave Functions, J. Chem. Phys. 121, 9313 (2004).
T. Hrenar, H.-J. Werner, G. Rauhut Accurate Calculation of Anharmonic Vibrational Frequencies of
Medium Sized Molecules Using Local Coupled Cluster Methods, J. Chem. Phys. 126, 134108 (2007).
molpro@molpro.net