Next: 43.2 Multi-level calculations
Up: 43 POTENTIAL ENERGY SURFACES
Previous: 43 POTENTIAL ENERGY SURFACES
The following options are available:
- NDIM=value
- The keyword NDIM=
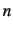 terminates the expansion of the PES after the -body term.
Currently, at most 4-body terms can be included, but the default is set to 3. Please
note, when you use NDIM=4 as a keyword for the SURF program, you
need to pass this information to the VSCF and VCI programs also. Otherwise
these programs will neglect the 4-body terms.
terminates the expansion of the PES after the -body term.
Currently, at most 4-body terms can be included, but the default is set to 3. Please
note, when you use NDIM=4 as a keyword for the SURF program, you
need to pass this information to the VSCF and VCI programs also. Otherwise
these programs will neglect the 4-body terms.
- NGRID=value
- Based on a coarse grid of ab initio points a fine grid will be generated from
automated interpolation techniques. The keyword NGRID= determines
the number of equidistant grid points in one dimension. NGRID= has to be an even number.
The default is currently set to 16.
Note that the number of grid points also controls the extension of the -dimensional potential
energy surfaces (see keyword SCALE) and thus influences many internal thresholds which are optimized to
the default value of NGRID. The number of grid points also determines
the number of basis functions in the VSCF program.
At present the maximum grid size is 20.
| Grid points |
14 |
16 |
18 |
20 |
| Surface extension |
4.30 |
4.69 |
5.05 |
5.39 |
- VAR1D=variable
- The SURF program reads the energy of electronic structure calculations
from the internal MOLPRO variables, e.g. ENERGY, EMP2,
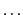 .
The internal variable is specified by the keyword VAR1D. Within the example
shown above, VAR1D=ENERGY would read the CCSD energy, while VAR1D=EMP2 would
read the MP2 energy, which is a byproduct of the CCSD calculation.
The default for the VAR1D keyword is the internal variable ENERGY.
.
The internal variable is specified by the keyword VAR1D. Within the example
shown above, VAR1D=ENERGY would read the CCSD energy, while VAR1D=EMP2 would
read the MP2 energy, which is a byproduct of the CCSD calculation.
The default for the VAR1D keyword is the internal variable ENERGY.
- DIPOLE=value
- Dipole surfaces can be computed for all those methods for which analytical gradients are available in
MOLPRO. For all methods except Hartree-Fock this requires the keyword CPHF,1 after the
keyword for the electronic structure method. The calculation of dipole surfaces is limited to
those multi-level schemes, for
which all variables VAR1D, VAR2D and VAR3D are set to the default, i.e. ENERGY
(see the section about the VMULT command). Symmetry is currently only implemented for
the 1D, 2D and 3D dipole surfaces. For 4D terms symmetry will automatically switched off at the
moment. The calculation of dipole surfaces effectively doubles the computation time for
surface calculations.
- SYM=value
- Symmetry within electronic structure calculations can be exploited by the keyword SYM=Auto.
Usually this leads to significant time savings.
By default this symmetry recognition is switched off as certain calculations may cause some trouble (e.g. local
correlation methods). Symmetry in electronic
structure calculations may not be mistaken for symmetry of the contributions to the potential
energy surface (see keyword MPG).
- SCALE=value
- The extension of the potential energy surfaces is determined from Gauss-Hermite quadrature points.
Using a fine grid NGRID=16 the surface stretches out to the NGRID/2
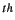 Gauss-Hermite point, i.e. 4.69, in each direction (see keyword NGRID).
As these values are fairly large within the calculation of fundamental modes,
a scaling factor, SCALE=
Gauss-Hermite point, i.e. 4.69, in each direction (see keyword NGRID).
As these values are fairly large within the calculation of fundamental modes,
a scaling factor, SCALE=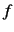 , has been introduced. A default scaling of 0.75 is used.
Increasing the size of the surfaces usually requires the calculation of further ab initio points
as the surface interpolation
is more stable for small surfaces.
, has been introduced. A default scaling of 0.75 is used.
Increasing the size of the surfaces usually requires the calculation of further ab initio points
as the surface interpolation
is more stable for small surfaces.
- FIT=value
- The iterative algorithm for generating potential energy surfaces is based on
a successive increase of interpolation points. The iterations are terminated once the interpolation
of two subsequent iteration steps became stable. The convergence threshold can be changed by
the keyword FIT=. There is currently just one control variable for the different
1D, 2D, 3D, and 4D iterations. The 4 thresholds are different but depend on each other.
Consequently, changing the default value (FIT=4.0d-2) will change all thresholds
simultaneously which keeps the calculation balanced.
- MIN1D=value
- The minimum number of coarse grid points can be controlled by the keywords
MIN1D, MIN2D, MIN3D, MIN4D. These 4 keywords determine the minimum number of
ab initio calculations in one dimension for each 1D, 2D, 3D and 4D surface.
The defaults are currently MIN1D=4, MIN2D=4, MIN3D=2, MIN4D=2.
- MAX1D=value
- The maximum number of coarse grid points can be controlled by the keywords
MAX1D, MAX2D, MAX3D, MAX4D. These 4 keywords determine the maximum number of
ab initio calculations in one dimension for each 1D, 2D, 3D and 4D surface.
The defaults are currently MAX1D=NGRID, MAX2D=NGRID, MAX3D=10, MAX4D=4.
- EXT12D=value
- Outer regions of the potential energy surfaces are determined by extrapolation rather than interpolation
schemes. An extrapolation of 15% in case of the 1D and 2D contributions to the potential (Ext12D=0.85)
and of 30% in case of the 3D and 4D terms (Ext34D=0.7) is currently used by default. For accurate
calculations extrapolation can be switched off by setting both keywords to 1.0. Changing
these keywords usually increases the number of ab initio calculations to be performed.
- SKIP3D=value
- As the number of 3D and 4D surfaces can increase very rapidly, there exist the possibility to neglect unimportant
3D and 4D surfaces by the keywords SKIP3D and SKIP4D. The criterion for the prescreening of the
3D surfaces is based on the 2D terms and likewise for the 4D terms the 3D surfaces are used. The neglect of
3D surfaces automatically leads to the neglect of 4D surfaces, as the latter depend on the previous ones.
By default prescreening is switched on, but can be switched off by SKIP3D=1.0 and SKIP4D=1.0.
- MPG=value
- Symmetry of the normal modes should be recognized by the program automatically. Only Abelian point groups
can be handled at the moment. Symmetry of the modes will be determined even if the NOSYM keyword
is used in the electronic structure calculations. In certain cases numerical noise can be very high
and thus prohibits a correct determination of the symmetry labels. This is denoted
by the label Err for these modes. In such cases symmetry should be switched off in the
calculation of the potential energy surfaces and the VCI calculations, because the results may
be corrupted. Symmetry can be switched off by using MPG=1.
- INFO=value
- INFO=1 provides a list of the values of all relevant program parameters
(options).
The following example shows the input of a calculation which computes energy and dipole surfaces
at the MP2/cc-pVTZ level and subsequently determines the anharmonic frequencies at the
VSCF and VCI levels. Hartree-Fock calculations will not be restarted and the .log-file
is directed to the scratch directory as defined by the $TMPDIR variable.
memory,20,m
geomtyp=xyz
geometry={mass
3
Water
O 0.0675762564 0.0000000000 -1.3259214590
H -0.4362118830 -0.7612267436 -1.7014971211
H -0.4362118830 0.7612267436 -1.7014971211
}
basis=vdz
logfile,scratch
hf
mp2
optg
{frequencies,symm=auto
print,low=50}
label1
{hf
start,atden}
{mp2
cphf,1}
surf,start1D=label1,dipole=1,sym=auto
vscf,dipole=1,combi=1
vci,dipole=1,version=3,combi=1
Next: 43.2 Multi-level calculations
Up: 43 POTENTIAL ENERGY SURFACES
Previous: 43 POTENTIAL ENERGY SURFACES
molpro@molpro.net
Sep 24, 2008